Störungen des Aminosäurenstoffwechsels Bekannt sind mehr als 70 angeborene oder erworbene Störungen des Aminosäurestoffwechsels (Aminoazidopathien). Sie sind jedoch insgesamt selten, d.h. etwa 1 auf 1000 Lebendgeburten. Aminoazidopathien lassen sich unterteilen in
Aminoazidurien treten auch bei Gesunden auf. Sie sind dann jedoch vorübergehend und mit einer erhöhten Aufnahme von Proteinen verbunden. Die Rückresorption der glomerulär filtrierten Aminosäuren geschieht in den proximalen Nierentubuli über Rezeptoren, die spezifische Aminosäuren bzw. Aminosäuregruppen erkennen und transportieren können. Störungen treten auf, wenn der Rezeptor gesättigt, kompetitiv durch andere Substrate besetzt oder in seiner Struktur bzw. Funktion gestört ist. Die eigentlichen Aminoacidurien können in primäre und sekundäre unterteilt werden. Die primären Aminoacidurien sind angeboren (inborn errors of metabolism). Sekundäre Aminoacidurien beruhen auf einer Organerkrankung (Leber) oder einer generellen tubulären Dysfunktion. Cystinurie Einen ersten Anhaltspunkt liefert der Nachweis von Cystinkristallen im sauren Urinsediment. Cystinkristalle sind farblos und treten als sechseckige Tafeln auf. Sie sind löslich in Salzsäure, nicht aber in Essigsäure. In bakterienhaltigem Urin werden sie rasch zerstört, für die Untersuchung muss also frischer Urin verlangt werden.
Abb. 1: Cystinkristalle Therapeutisch steht eine Steigerung des Urinvolumens im Vordergrund. Angestrebt wird eine Flüssigkeitszufuhr von 4-8 Litern täglich. Da die Wasserlöslichkeit von Cystin bei einem pH von mehr als 7.5 rasch zunimmt, kann auch eine Alkalisierung des Urins mit Natriumbicarbonat oder Acetazolamid versucht werden. Generalisierte Aminoazidurien
Cystinose Alkaptonurie
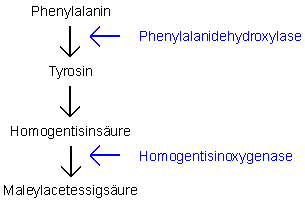
Abb. 2: Stoffwechsel des Phenylalanins. Phenylalanin ist der Ausgangspunkt lebenswichtiger Verbindungen Es kommt zu einer vermehrten Ausscheidung von Homogentisinsäure im Urin, aber auch zu einer Homogentisinanhäufung im Gewebe. Wenn sich Homogentisinsäure an Kollagen bindet, entsteht eine blaugraue Pigmentierung, die als Ochronose bezeichnet wird. Bevorzugte Stellen sind wenigdurchblutete Gewebe wie Ohren- und Nasenknorpel, Skleren, Bandscheiben. Klinisch äussert sich die Erkrankung durch die ochronotische Pigmentierung und eine Arthritis mit gichtähnlichen Schmerzanfällen sowie degenerativen Veränderungen an Gelenken und Wirbelsäulen. Diagnostisch sind Dunkelfärbung des Urins bei längerem Stehenlassen und die Messung der Homogentisinsäure im Urin.
Phenylketonurie Die Krankheit wird in der Schweiz durch das allgemeine Screening von Neugeboren erfasst. Andernfalls kommt es ohne Behandlung, die innerhalb von 30 Tagen nach der Geburt einsetzen muss, zu einer Anhäufung von Phenylalanin und seiner Metaboliten. Dies führt innerhalb des ersten Lebensjahres zu schwerer mentaler und psychomotorischer Retardierung, sowie zu Hyperaktivität, Tremor und Krampfanfällen. Die Behandlung besteht in einer Diät, bei der ein grosser Teil der Proteine durch ein phenylalaninarmes Aminosäurengemisch ersetzt wird. Homocystinurie Die Anhäufung von Homocystin führt zu einer Störung der Kollagenbildung.
Um einen möglichen Schaden zu verhindern, müssen Störungen des Aminosäurenstoffwechsel so rasch wie möglich nach der Geburt diagnostiziert werden. Allerdings sind diese Krankheiten selten und die Symptome oft unspezifisch. Abklärungen sind indiziert bei ständigem Erbrechen, fehlender Entwicklung, neurologischen Symptomen, besonders wenn sie mit Änderung der Ernährung einhergehen. Diagnostisch werden zur Bestimmung von Aminosäuren folgende Methoden eingesetzt:
|
||||||||||
|
21.11.2000 / hpk |